Nøkkelen til denne syntesen burde være - etter min mening - hvordan man genererer de to hydroksyfunksjonene med anti stereoselektivitet med utgangspunkt i minst ett achiralt reagens. Mesteparten av tiden vil du prøve å bygge opp chiralitet de novo ved å bruke chirale hjelpestoffer, katalysatorer eller reagenser, da disse metodene ofte er mer kostnadseffektive enn å anskaffe en spesifikk chiral reaktant - det åpenbare unntaket er hvis en forbindelse eksisterer i det naturlige chirale bassenget som du enkelt kan bruke (som egentlig ikke er tilfelle her). Etter å ha identifisert vårt viktigste problem, kan vi vende oss til de andre:
- vi trenger å generere en δ-lakton
- vi trenger å danne en $ \ ce {CC } $ obligasjon, helst på en chiral måte, til en $ \ ce {C6} $ kropp (biten av det endelige produktet som mangler i reaktanten).
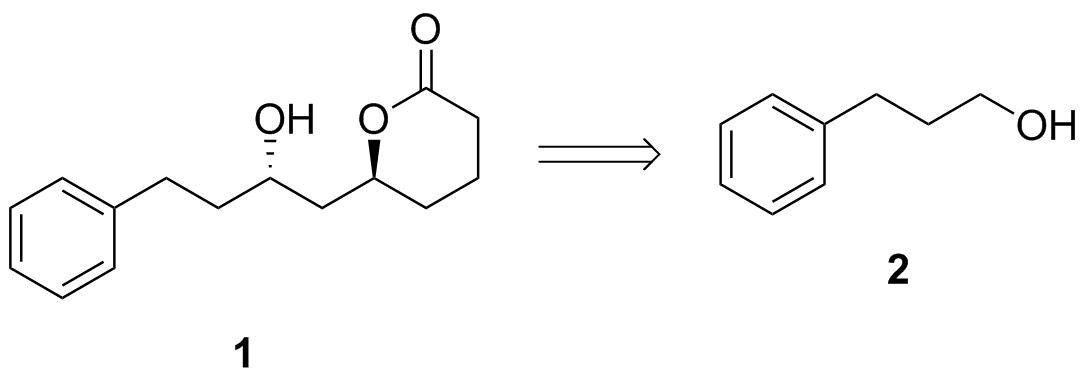
Skjema 1: Generell retrosynteseide.
Etter min mening er δ-lakton et ikke-tema. Hvis du har en ester med åpen kjede som 3 , kan du sannsynligvis danne seksleddet lakton ganske enkelt ved $ \ ce {K2CO3} $ - mediert transesterifisering. Praktisk talt alle litteraturmetoder for å syntetisere estere bør også overveiende gi δ-lakton, spesielt siden den ugunstige ζ-laktonen er alternativet. (I det usannsynlige tilfellet at den dannes, kan den enkelt transesterifiseres som nevnt.) Jeg vil sette dette trinnet sist.
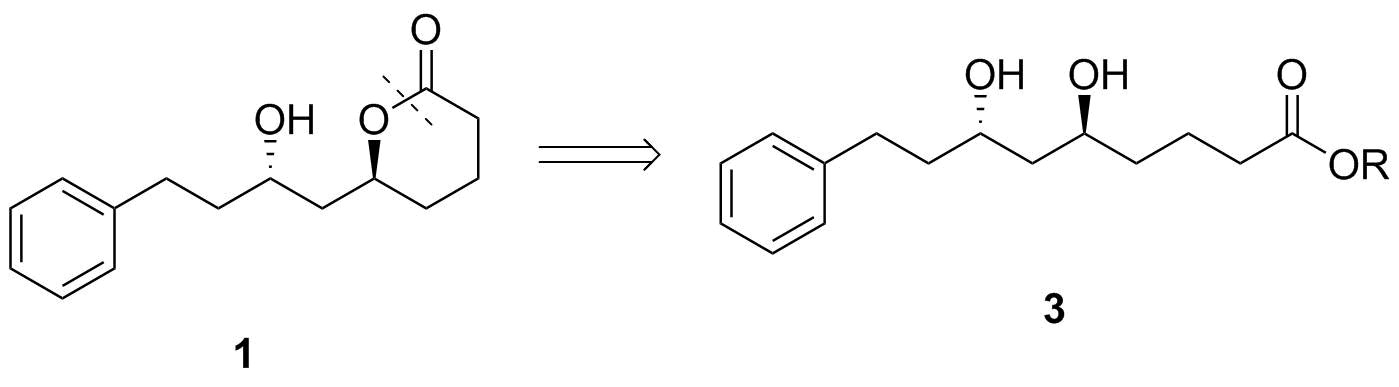
Ordning 2: Transesterifiseringstrinn.
Vi har nå 3 , en anti -konfigurert 1,3-diol. Når jeg ser dette, hopper tankene mine umiddelbart til Evans-Tishchenko asymmetriske reduksjon. Dermed kan vi komme dit fra en av de to mulige β-hydroksyketonene 4 eller 5 . For at dette skal fungere best, vil jeg at karboksygruppen skal maskeres som en beskyttet alkohol her, jfr. 6◄.
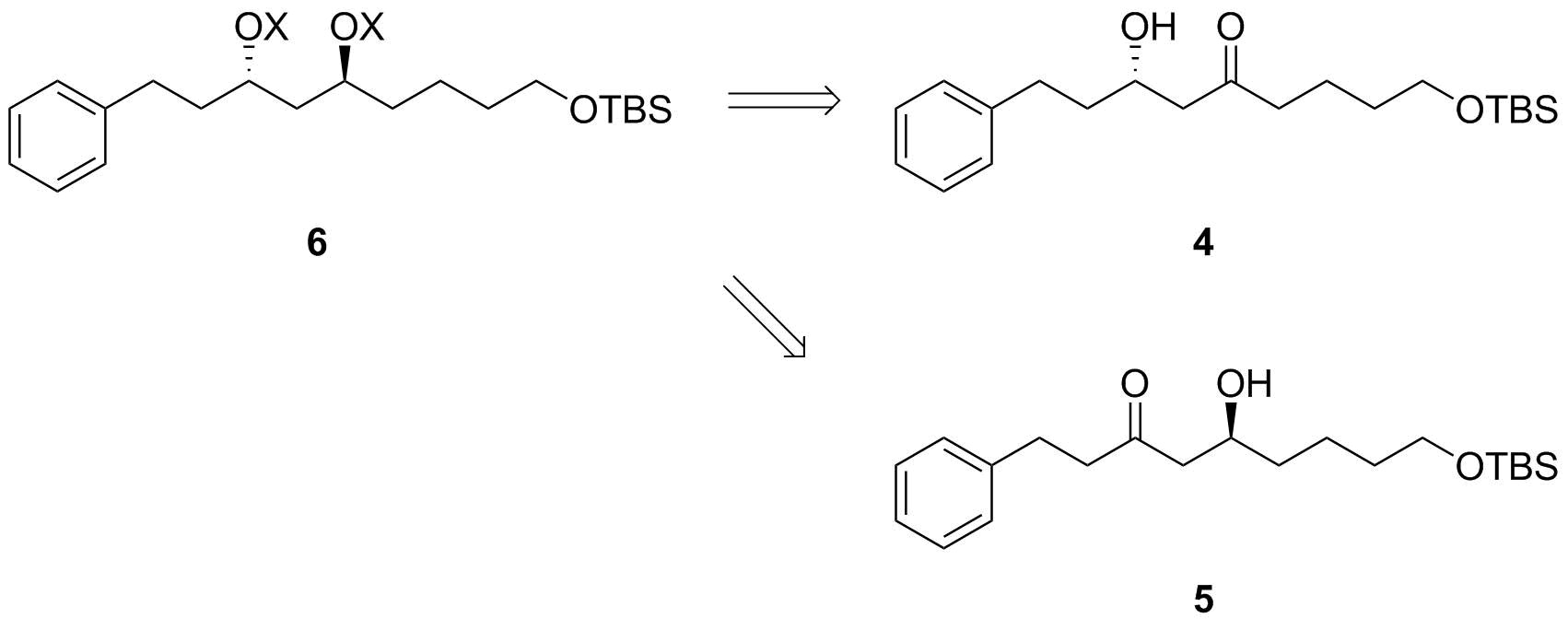
Skjema 3: Evans-Tishchenko-trinn. $ \ ce {X} $ ville være $ \ ce {H} $ for den nylig dannede hydroksygruppen og $ \ ce {CH3CH2CO - {}} $ for den induserende.
Begge disse β hydroksyketoner kan genereres fra aldolreaksjoner; Imidlertid antyder bare keton 4 at det lett kan nås fra ønsket reaktant 2 . Hvis vi ønsket å syntetisere 5 , ville vi trengt et ekstra karbonatom på 2 som vi ville trenge å introdusere i en ytterligere reaksjon. Veien til 4 krever imidlertid bare selektiv oksidasjon til aldehydet - min personlige favoritt er Dess-Martin – oksidasjon, men du bør nok sjekke alle de andre som foreslår seg selv ( Swern og slektninger, TEMPO, Ley-Griffith, osv.) som man kan være overlegen over de andre på grunn av at laboratorieåndene dine liker det.
Til slutt forteller dette oss også at aldoltilsetningen for å generere 4 fra 7 og 8 skal fortsette på en stereoselektiv måte som gir ( S ) -konfigurert produkt overveiende. Igjen, mange metoder er kjent, og jeg vil foreslå en Paterson aldol-reaksjon. Analyse av relevant litteratur (dessverre fant jeg ikke et nettsted, så det er en sitering) avslører at $ \ ce {(-) {-} Ipc2BOTf} $ og Hünig-basen skal gi ( S i >) produkt fortrinnsvis. [1]
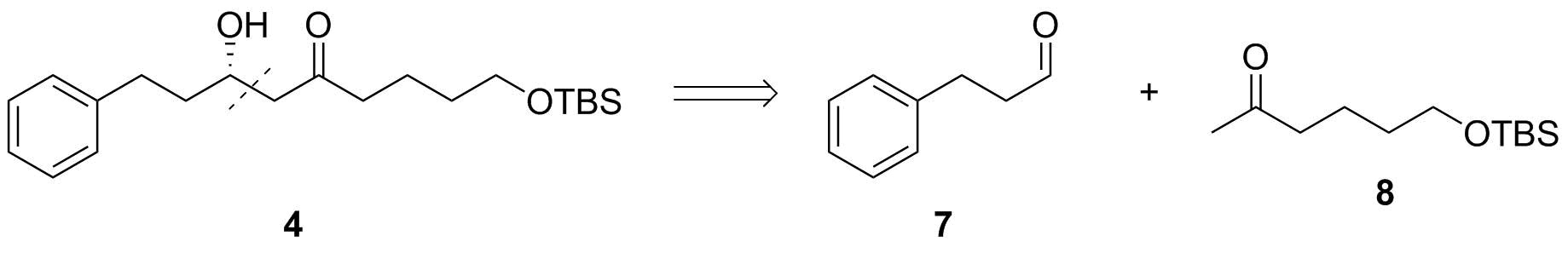
Skjema 4: Endelig aldolfrakobling.
Dette fører oss til følgende fremover syntese:
$$ \ ce {\ mathbf {2} -> [DMP] \ mathbf {7} -> [(-) {- } Ipc2BOTf] [(iPr) 2NEt, \ mathbf {8}] \ mathbf {4} -> [SmI2, EtCHO] \ mathbf {6} -> [1) TBSOTf; 2) PPTS] [3) DMP; 4) {Pinnick}] \ mathbf {3} (R $ = $ H) -> [DCC, DMAP] [K2CO3] \ mathbf {1}} $$
Referanse :
[1]: A. S. Franklin, I. Paterson, Contemp. Org. Synt. 1994 , 1 , 317–338. DOI: 10.1039 / CO9940100317.